��������ˎ����һ����ìF�����\�g�������a��Դ�������w�Ȳ������ڼ������\�ࡢ�ί����A�����������ӣ��M�x��Ҳ���Q�����\�gˎ������硢���wˎ������ο�¡���w�����wżˎ�ADC�����p�خ��Կ��w��Fc �ںϵ��ס����wƬ�Ρ����¡���w�ȡ��c���W�ϳ�ˎ����ȣ���������ˎ����������������Y�����s�������^����Ĥ���oˎ�����ͺ������w�Ƚ�������c���S�����\�g��Ѹ�Ͱlչ����������ˎ���ѱ��V�������ί��[�������������Լ����ʹ��x�Լ����ȶ�N������Ȼ������ˎ�����a���g�ͱ����������^������c�������ˎ�����������F����ԭ��Immunogenicity����
����ԭ�ԣ�Immunogenicity���Ǯ��ί���aƷ����ijЩ�������ί�ˎ��r�����C�w�R�e��������|����ԭ�����a�������ߑ���Ȼ�������ί��Ե����@һ���ߑ��������Dz���Ҫ����A�ڵģ����ڴˑ����^���пɌ��C�w�a����ˎ���w��Anti-drug antibodies��ADAs����������ˎ���w���кͿ��w�����ߏͺ���ȣ�ADA�Ĵ��ڿ�Ӱ�ˎ�������W���ԡ�ˎ�������W����������ʹ��ˎ��ԭ�еį�Ч���ͻ�ȱʧ��������Щ���ߑ����c�C�w��Դ�Ե��a���������߷������t��������ص��R����ȫ���}��
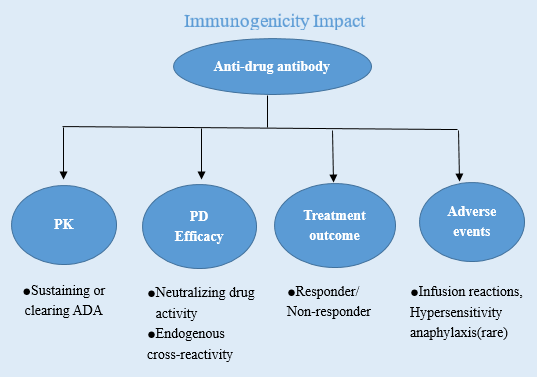
�Dһ ����ԭ�Ե�Ӱ�
���ǻ��ڴˣ� ����ԭ�Ե��u��ؽ�����P���gҎ���ļ�����֧�֡�
2007��W��ˎƷ�O�ܙC����EMA��������ˎƷί�T�� (CHMP) �l������ԭ���u����ָ��ԭ�t����δ����ήa������������������Ԕ��Ҏ������2017��EMA�l����ԓָ��ԭ�t����K�汾��Final Version�������ԛ]�г��F���g�����ϵ�Ԕ��Ҏ����EMA؞�����m��Ŀ��ԭ�t��fit to purpose, FTP����Ҫ�����k���x����ķ�����푑���M��O��Ҫ���������`���ԡ�
����FDA�t��2014���״ΰl��������ԭ���u��ָ��ԭ�t�����нo������ԭ�Ե�Ӱ����غ��u�r���ԡ�����2019��l��������ԭ���u��ָ��ԭ�t��Immunogenicity Testing of Therapeutic Protein Products ��Developing and Validating Assays for Anti-Drug Antibody Detection Guidance for Industry ���Ќ�������ԭ�ԡ����x���ί��Ե��aƷ�����������P���a�����ߑ�����T�l�������P�R�������¼��ăA��
����ˎƷ�O�������֣�NMPA����2020��08��24�հl���ˡ�ˎ������ԭ���о����gָ��ԭ�t��������Ҋ�壩��������2021��03��29�հl����ʽ�汾���係�w���u�������cFDA��ͬ���c���е�FDA��EMAָ��ԭ�t������ͬ��NMPA��ʽ��t��ֿ��]���˷��R�����R���о�������ԭ�ԙz�y�����IJ�e���@һָ��ԭ�t�ij��_�����������ԭ�����P�о��ṩ�����P��Ҫ��Ҏ����ָ����
����ADA�ęz�y��NMPA��FDA��EMA�ęz�y�Q����һ�£����߾��ᳫ���Ӽ����u�����ԣ�
��1���Y�xԇ�����Иӱ��M�У��Y�x��������Ԙ�Ʒ��
��2���_�Cԇ��������Ԙӱ��M���خ��ԵĴ_�Cԇ�_����Ԙӱ���
��3���ζ�ԇ���Ѵ_�����w��ԵĘӱ��M�еζ�ԇ�_���������������������ԇ���w�M���кͻ��ԙz�y��
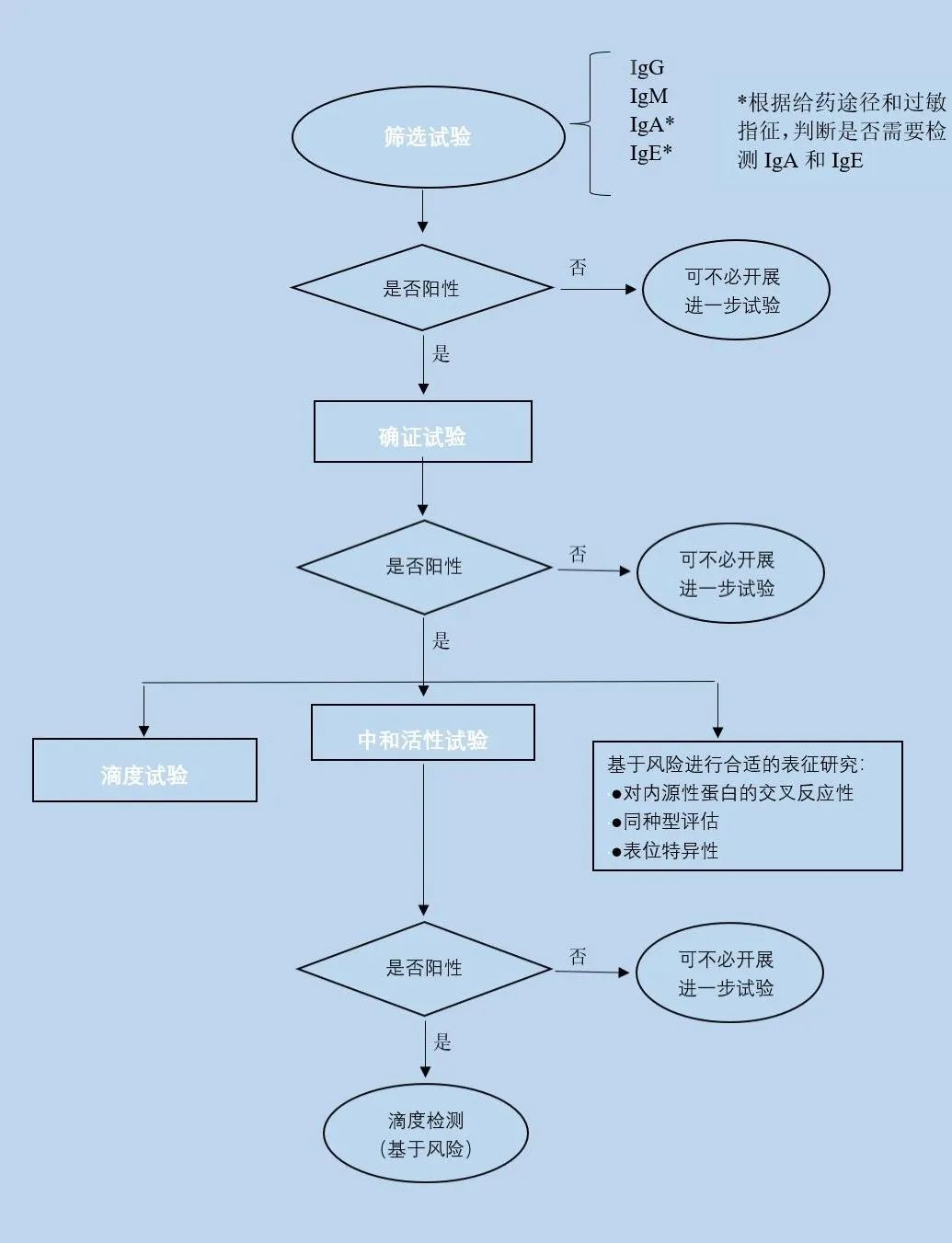
�D�� ADA�z�y�ķּ��u������
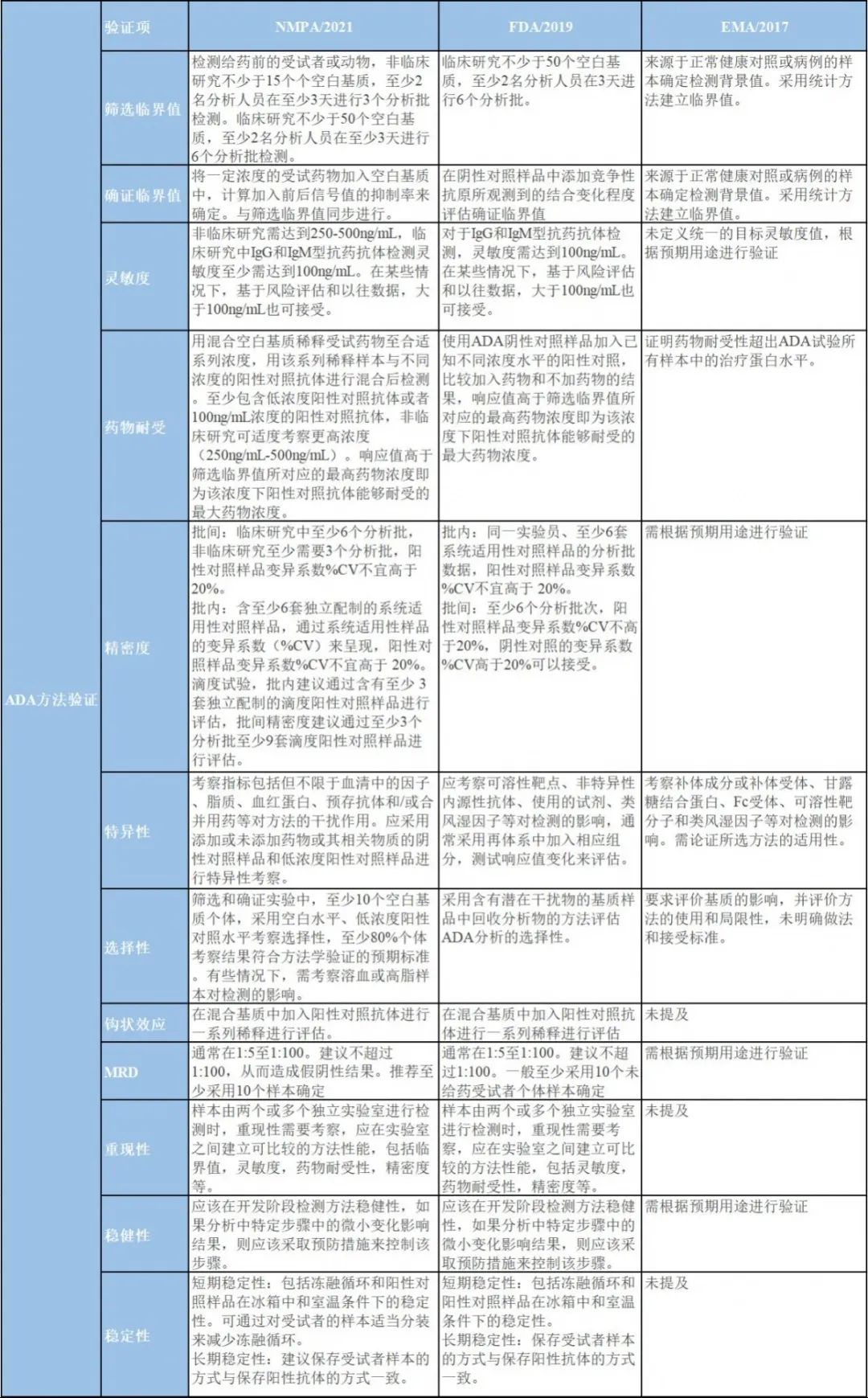
�D�� NMPA��FDA��EMAָ��ԭ�t����C�ă��ݺͽ��ܘ˜ʮ�ͬ�c
���ڿ�ˎ���w�z�yᘌ���ͬ�ęz�y������y�ȿ��Բ��ò�ͬ�ķ����̓x�����������������ڣ�ֱ�ӷ��������;���Y�Ϸ��ȡ����y�ęz�y�x����ø�˃x��MSD�ȣ�Ŀǰ����MSD��늻��W�l�⣩ ƽ�_�Ę�ӷ���ADA�z�y�����ķ��������ڰ����f���tˎ�����yԇ����ƽ�_�����д�����Ƶęz�y�O��Ͷ��������������z�y�������wˎ�������W(PK)��ˎЧ�W(PD)����ˎ���w(ADA)���кͿ��w(NAb)���������־��(Biomarker)�z�y�ȣ��ɸ����͑������ṩ���Ƶķ����W�_�l����C������ӱ��z�y���գ��gӭȫ�����ص�����ǰ��������ԃ��

�����Y��
��1�ݡ�ˎ������ԭ���о����gָ��ԭ�t(������Ҋ��)��NMPA, 2020.08
��2�ݡ�ˎ������ԭ���о����gָ��ԭ�t��,NMPA, 2021.03.
��3��Immunogenicity Testing of Therapeutic Protein Products �� Developing and Validating Assays for Anti-Drug Antibody Detection Guidance for Industry, January 2019.
��4��Guide line on Immunogenicity assessment of biotechnology-derived therapeutic proteins (EMEA/CHMP/BMWP/14327/2006 Rev 1), 1st Dec 2017.



 ��W���� 34012302000810̖
��W���� 34012302000810̖