�|�ӱ����Ƅ���PPI������Ҫȱ�c���£���i��������������ģ�һ����Ҫ�Ƃ���c���Ƅ����@��Ӱ���θ�ſ����ʣ��@Ҳͬ�r�����ˎ�����Õr�g��ʲô���r����l���@���IJ�ͬ����ii��ˎ�������W���ڂ��w�������@Щˎ����Ҫ�ɸ�ˎ����xøCYP2C19���x�����@ʾ�z�����B��; ��iii��������ڵ�����Ч�����ܳ��m24С�r����ˣ�PPI���ܳ������ҹ�g�����;��iv���@Щˎ����о�������Ч���ã�ͨ������������Ч�������@�ġ�����@Щ���}�õ���Q��δ�M����t�������p�٣��M���@Щ�����ˎ������������cθ�����P���������ί��x���о��l�F��⛸��������������P-CAB���İlչ��ϣ����������ѵĽ�Q������
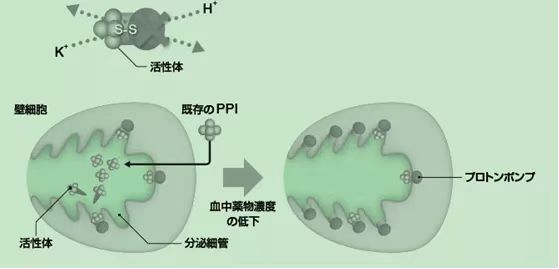
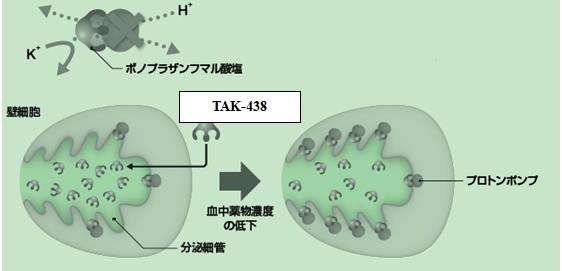
P-CAB����ͨ�^����H+��K+ -ATPø���cPPI�����õIJ�ͬ�C�ơ�PPIͨ�^�cø�γɹ��r�I�������������H+��K+ -ATPø����P-CABͨ�^�cø��ǻ��K+�Y��λ�c�ϵ�K+�x�Ӹ��������������H+��K+ -ATPø��Ҋ�D1��
�D1 PPI��TAK-438�����ÙC�Ʊ��^
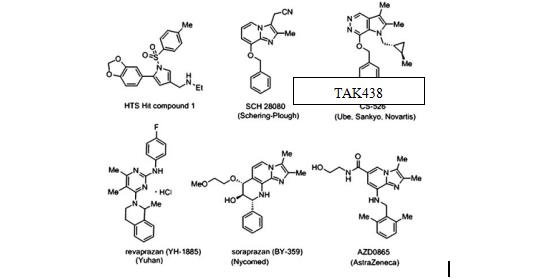
��20���o80����ԁ���һЩ��ˎ��˾ԇ�D�_�lP-CAB���������������Ĺ�Ч�ζ���[1-2]��ͨ�^̽�����о����l�F����K�������Ƅ����˷�PPI�ľ����ԣ�ͬ�r��������ǰ�_�l��P-CAB���F���IJ����Ч�ʺ͝��ڵĸζ��ԡ�
���˰l�Fһ�N�µ������Ƅ����о�����2003���ԁ����ø�ͨ���Y�x��HTS�������u�r��560 000�N������ɹ��b����������������H+��K+-ATPø���ƻ��ԣ��������ԗl���¾������õĻ��W�����ԡ�ԓ��������������������A�ԵĽY��;������λ��1-λ����5-λ�ķ��h���һ�-����-�����܈Fλ�������h��3-λ�����⣬�����еͷ������ͽY��׃��������ă��c�����仯�W�Y���Ǫ��صģ����Ҍ��������Ƅ���ǰ��δ�еġ��о��ߌ����л�����1������m�о�����������HTS Hit Compound 1�����w�Y��Ҋ�D2��
�D2 HTS hit compound 1�����������P-CAB���W�Y��
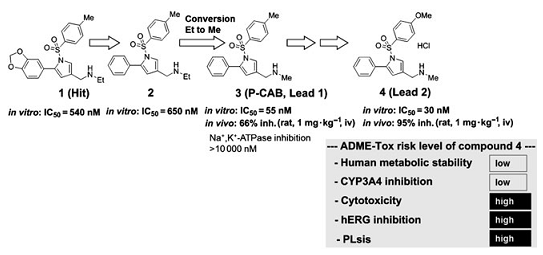
�о����u�������л�����ĝ������������������M���˺ϳ��о������u���˘�Ч�Pϵ��SAR����Ҋ�D3������1��[3]�����ȣ��ϳɻ�����2���C�������h5λ�ϵĉA��SAR������ā��Y������ʹ�û�������Ե��ԾS�֡���������3-λ�ϵ�N-�һ�������׃��N-���������r����������w�����ӳ��^ʮ�������⣬������3��1mg▪kg-1���o�}�ȣ��ڴ���������66��������ڡ��cNa+��K+-ATPø��ȣ���߀�@ʾ����H+��K+-ATPø�ĸ��x���ԡ����⣬�����W�о�����H+��K+-ATPø������K+�ĸ��������ƣ�����ԓ��������P-CAB����ˣ��S���䌦ø�Ļ������ӌ����@������������ƣ��Լ��ߵ�ø�x���ԣ����������������P-CAB��һ���������������Ƶ����ú��x�
�D3 HTS hit compound 1 ��������
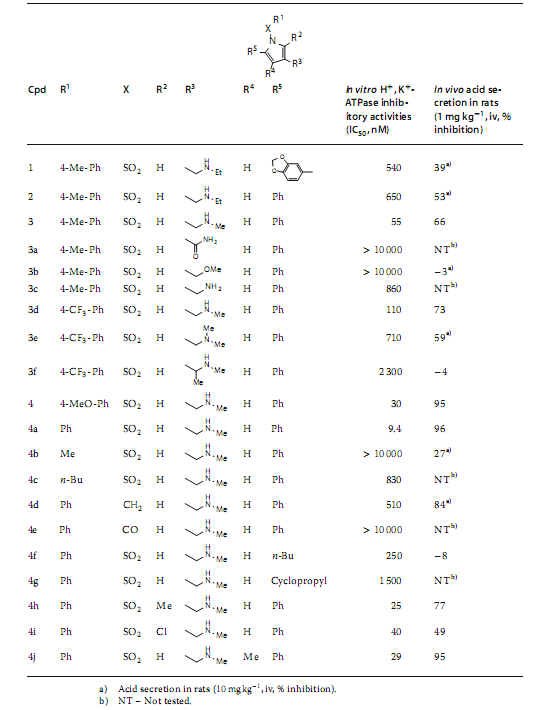
��1 HTS hit compound ��SAR��^��
�˺��x����3�����Ȍ������lead 1�������C���о���SAR����ˣ�N-����������3λ��R3���֣����F���e��Ч�����ƻ��ԣ���1λ��X��R1���֣��������c������ֱ���B�ӵķ���h��ֱ���B�ӵķ��h��5λ��R5���֣�������ġ����⣬������2λ��R2���֣���4λ��R4���֣���ȡ�����r�����ԛ]���@�����ƣ�Ҋ�D3����
��SAR�^���У�Ҳ�������õ��������W���|�����w���x�����ԵĻ�����4��1mg▪kg-1�����@ʾ���m������90�����ƣ����������������ã�95�����ƣ����M�ܻ�����4�@ʾ���ЎN��������ADME-Tox���|�����缚�����ԣ�hERG���ƺ����ߵ���֬����PLS���L�U�������Ա�����һ�N���ڵ��Ȍ������
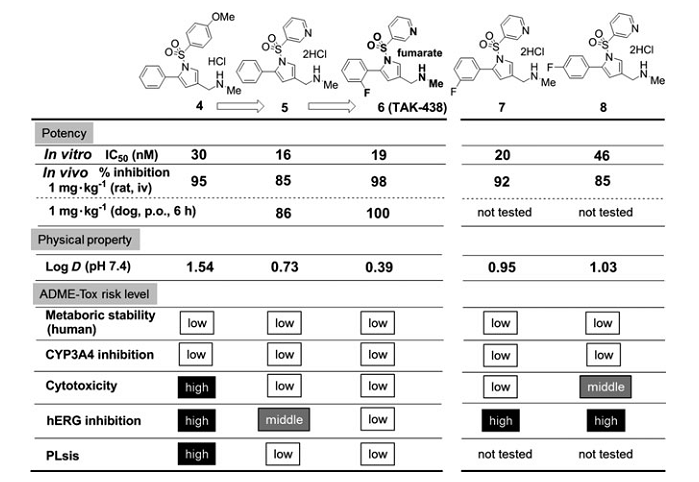
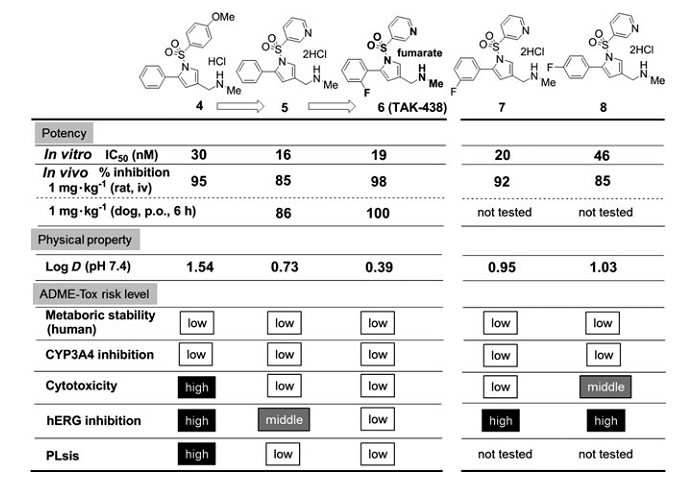
�����µĺ��x������ă�������ֱ�Ӻ����_�����ڻ����lead 2���Ȍ�����һֱ�ڳ��m�M�С�ADME-Tox�����ęz��ͨ�������^�ͻ��ԵĻ������M�С��l�F���H�y����log Dֵ���w�⼚�����Ԕ���֮�g����������������P�ԡ����⣬߀�^�쵽�y����log Dֵ��hERG����֮�g���p���P�ԣ����wҊ�D4��
�D4 ����������Ĝy��log Dֵ���w�ⶾ�Ԕ���֮�g�����P��
�����@Щ�l�F���о����܉��γ�һ�N���O����ͨ�^�@��������logDֵ�������@�����������������ADME-Tox�ֲ��������ڃɂ��ش��}��һ�����}�Ǻϳɷ�������������O�Ի��F�M�������h��1-λ����Ҫ�������^�͵Ļ��W�����ԡ���һ�����}�ǵ�log D��������w�Ȼ����@�����͡��_�lһ�Nʹ��15-��-5�������ӄ����ºϳɷ�����Q��ǰ�����}���Ķ������������h��1λ������N�O�Ի��F���о��߇Lԇʹ�ýY�����w����Ч��/�w�ⶾ�Ԕ���֮�g���Pϵ��Q��һ���}���������������h1λ�ϵĘO�Ի��F��λ�ã�Ȼ���������h��5λ����K�_����3-��ऻ��������ȡ����֮һ�������log Dֵ[4]��
���R����Z������TAK-438��������xˎ��ĽY��
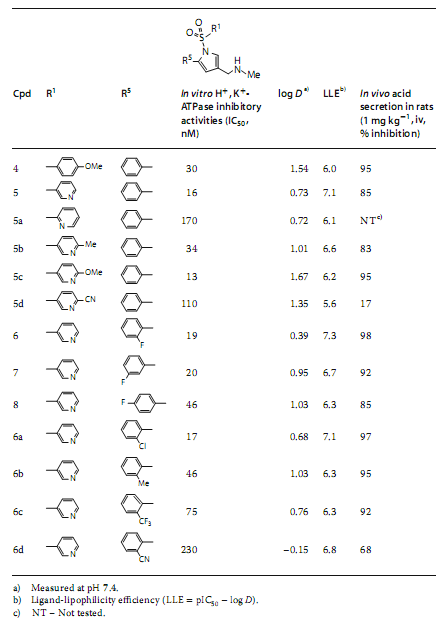
���OӋ�Ļ������Ӌ�����w�H֬��Ч�ʣ�LLE = pIC50-logD��ֵ����ˎ�������Ե�ָ�ˣ����wҊ��2��
�D5 ͨ�^�����x����6��TAK-438�����R����Z������
��2��lead compound 4��������������
�����u�r���N�ϳɻ�����ĽY������3-��ऻ�����R1���ֵõ�������5��Ҋ�D5�������@ʾ0.73�ĵ�log Dֵ����߀����˼������Ժ���֬���e���L�U�����]���@�������w��Ч�ʡ����⣬���Ќ�2-F-Ph���F����R5���ֵĻ�����6�a���^�͵�log Dֵ��0.39��ͬ�r���֏���H+��K+-ATPø���ƻ��ԡ����⣬���ڴ������@ʾ������Ч�������ڹ����@ʾ���@���Ŀڷ����ԡ�������6�����w�H֬��Ч��LLE�u�rֵ�^�ߣ���ֵ��7.3������hERG���Ƶ��L�UҲ��͡���ˣ�ԓ�������m���pС���w��ADME-Tox�������Pע��
���錦�ȣ����в�ͬλ�õ�Fԭ�ӵĻ�����7��8�@ʾ�Ȼ�����5���ߵ�logDֵ��Ȼ�����@Щ����������Ч�ʺ�ADME-Tox���|�������@���㡣
��ˣ��о����x����6��TAK-438�����R����Z�����������_�l�ĺ��x�����Ҋ�D5����
�ں��m��ԇ��У�TAK-438���H���R��ǰ�о��У�ͬ��Ҳ���R��ԇ��гɹ���ȡ���������ĽY�������R���о��У�TAK-438�������õİ�ȫ�Ժ����õ������ԡ�
1��������Ҫ����J��nos Fischer and Wayne E.Childers�����ĕ���Successful Drug Discovery Volume 2�еĵ�10����Discovery of Vonoprazan Fumarate (TAK-438) as a Novel, Potent and Long-Lasting Potassium-Competitive Acid Blocker��
2��ԓ���H�����P�߂��ˌ�����ˎ�_�l��һЩ�뷨�����⣻�gӭ��λ�ώ����uָ����
1.Parsons, M.E. and Keeling D.J. (2005) Novel approaches to the pharmacological blockade of gastric acid secretion. Exp. Opin. Investig. Drugs, 14, 411�C421.
2.Kahrilas, P.J., Dent, J. and Lauritsen, K.(2007) A randomized, comparative study of three doses of AZD0865 and esomeprazole for healing of refluxl esophagitis. Clin. Gastroenterol. Hepatol.,5, 1385�C1391.
3.Nishida, H., Hasuoka, A., Arikawa, Y. et al. (2012) Discovery, synthesis, and biological evaluation of novel pyrrole derivatives as highly selective potassium-competitive acid blockers. Bioorg. Med. Chem., 20, 3925�C3938.
4.Arikawa, Y., Nishida, H., Kurasawa, O. et al. (2012) Discovery of a novel pyrrole derivative 1-[5-(2-fluorophenyl)-1-(pyridin-3-ylsulfonyl)-1H-pyrrol-3-yl]-N-methylmethanamine fumarate (TAK-438) as a Potassium-Competitive Acid Blocker (P-CAB). J. Med. Chem., 55, 4446�C4456.
����ǢՄ��������
ϵ��ʽ��13635699896����̖ͬ��
����ǢՄ��������
ϵ��ʽ��15156091013����̖ͬ��
�gӭ����ȫ�����صĺ�������W�R��˾���^������

 ��W���� 34012302000810̖
��W���� 34012302000810̖